
Phylogenetics, Systematics, and Bioinformatics
Lab Assignment
In this lab you'll be exploring the classification of and evolutionary relationships among some odd-looking fish: four species of flatfish (the engraving below shows a few of the species you'll be studying, along with a few more...look for the flatfish hiding on the seafloor!).
Beginning with a published phylogenetic tree that was constructed based on painstaking analyses of flatfish morphology, you will then explore how DNA sequences can be used to evaluate these relationships. First, you'll manually compare some short DNA sequences; then, using full gene sequences, you will use two online tools called Clustal Omega and ClustalW2, developed and hosted by the European Bioinformatics Institute, to conduct a multiple sequence alignment and to derive a type of phylogenetic tree known as a cladogram.
In order to complete this assignment, you will need to:
By the end of this assignment, you should be able to:
1. Use a phylogenetic tree as a hypothesis in order to generate a testable prediction.
2. Align and compare short example DNA sequences and calculate pairwise alignment scores, in order to evaluate the hypotheis.
3. Use an online bioinformatics resource to perform a multiple sequence alignment and comparison and to constructo a phylogenetic tree.
4. Identify similarities and meaningful differences between two phylogenetic trees.
Wood engraving of several flatfish species, from Alfred Edmund Brehm, Illustrirtes Tierleben (1864-1869). [Public domain], via Wikimedia Commons.
Notes
This assignment is based in part upon Gass G, Welsh E. 2011. Lab 7: Phylogenetic trees. BIOL 1010, Department of Biology, Dalhousie University.
In this assignment, you'll explore the evolutionary relationships between several different species of fish. The fish you'll meet are commonly known as "flatfish", and the proper classification of these fish has been a work in progress for decades. In this first section of the assignment, you'll watch the video called "Meet the Flatfish", and work with a phylogenetic tree that was generated based on detailed observation of the morphology (the structure of the body and its parts) of the several groups that make up the larger group known as the flatfish.
A phylogenetic tree is a visual hypothesis about the relatedness of the species or groups that it includes, and like any other hypothesis it can be used to generate predictions. When interpreting a phylogenetic tree, it's important to remember that the more closely related two species are, the shorter the distance to their most recent common ancestor on the phylogenetic tree. For example, in the tree below, species A is more closely related to species B than to species C.
Watch Meet the Flatfish, and use the video plus the images on this page to answer the questions in this section of the assignment document.
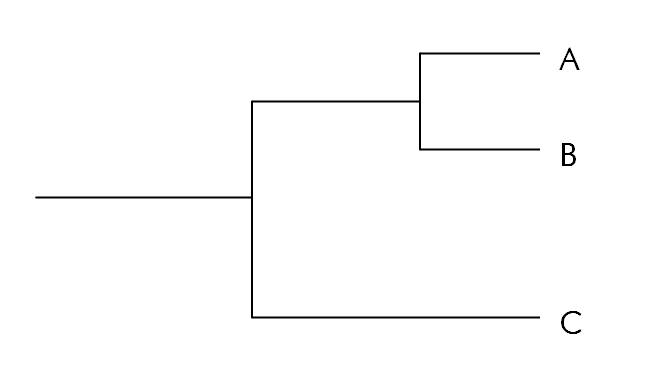
Figure 1. Sample phylogenetic tree showing the evolutionary relationships of species A, B, and C.
The arrangement of the flatfish families in the phylogenetic tree shown in the video (and reproduced in Figure 2 below) is based upon Chapleau's 1993 study, which generated a phylogenetic tree arranging the flatfish families based on extremely detailed morphological study of the members of those families.
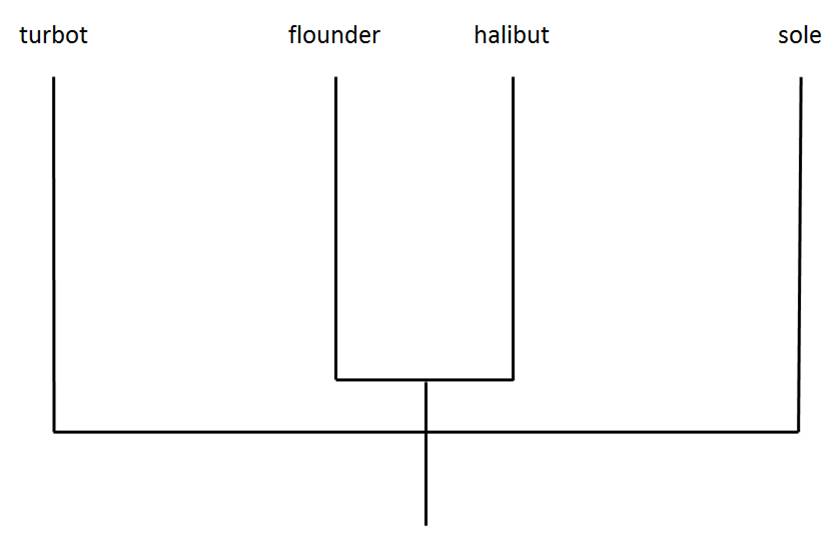
Figure 2. Phylogenetic tree displaying evolutionary relationships among four flatfish species, based on morphological evidence. Adapted from Chapleau (1993).
In Figure 3 below, you will see the four representatives of the order Pleuronectiformes that you learned about in the video, and you will also see a representative of another order, the Salmoniformes. Recall that 'order' is the taxonomic level below 'class'. The orders Salmoniformes and Pleuronectiformes belong to the same class, the Actinopterygii (the "ray-finned fishes"). All of the members of class Actinopterygii share several major morphological features such as the structure of their scales (Hart and Reynolds, 2002). Similarly, all of the members of each order within the class share certain features that distinguish them from members of other orders, and all of the members of each family within an order share certain features that distinguish them from members of other families.
Click on any of the fish to open up a larger version of the figure.
 .
.
Figure 3. Representatives of two orders of ray-finned fish. Solea senegalensis, Platichthys stellatus, Psetta maxima, and Hippoglossus hippoglossus belong to the order Pleuronectiformes; Salmo salar belongs to the order Salmoniformes.
Recall once more that a phylogenetic tree is a hypothesis about the relatedness of the groups shown on the tree. Morphological data were traditionally used to generate phylogenetic trees, but in recent years it has become possible to compare DNA and protein sequences between groups, and to use this data to generate phylogenetic trees. Sometimes the relationships proposed by a tree constructed using molecular data are the same as the relationships proposed by a tree constructed using morphological data, and the molecular data provide support for the morphological data. Other times, molecular data help to reveal aspects of the relationships between species that were not apparent when using morphological data alone.
Because a phylogenetic tree is a hypothesis, we can use the tree to generate a testable prediction. If the species on the tree in Figure 2 are related in the way that this tree suggests that they are based on their morphology, then we might expect the species to show these resemblances at the molecular (DNA and protein) level. In the next section of this assignment, you will test a prediction about the molecular similarities of your fish species. First, you will use the morphology-based phylogenetic tree from Figure 2 to generate a prediction (see the related question in the assignment document).
See the assignment document for full instructions for this part of the lab.
Figure 1: created by Gillian Gass.
Figure 2: adapted from Chapleau F. 1993. Pleuronectiform relationships: a cladistic reassessment. Bulletin of Marine Science. 52: 516-540.
Figure 3 (alphabetical order by common name):
References
Hart PJB, Reynolds JD. 2002. Handbook of fish biology and fisheries, vol. 1. Wiley-Blackwell.
In this section of the assignment, you will learn to use two powerful online tools for comparing DNA and protein sequences. There are many different software tools used by evolutionary biologists. In today's lab, you will use a set of web-based tools called Clustal Omega and Clustal W2, hosted by the European Bioinformatics Institute.
In the previous sections of this assignment, you have analysed a phylogenetic tree that is based on morphological features of the taxa being compared. You have also manually compared short example DNA sequences from each fish species to determine pairwise alignment scores, a mathematical representation of the similarity between two species based on a particular DNA or protein sequence. As you compared the DNA sequences, you probably noticed that this process is both tedious and error-prone - and your sequences were only twenty-three base pairs long! Biologists routinely compare much longer sequences, and often make comparisons between a very large number of taxa. Computers are ideal for performing tasks that humans find tedious and error-prone, and in this section of the assignment you'll see some of the information-management and calculation tools that are used in bioinformatics work.
To complete this section of the assignment, you will need this assignment document, the file called "FishSequences.txt", and the ClustalInstructions PDF. If you click on the .txt file and it automatically opens in a new tab of your browser instead of downloading, then just highlight the whole area of text and copy it (Ctrl-C on a PC or Command-C on a Mac).
The "FishSequences.txt" file contains complete actual DNA sequences for the four flatfish species for the gene COX1, which codes for one of the subunits of the mitochondrial enzyme cytochrome oxidase. A section of the COX1 gene sequence was selected as the "Barcode of Life" in the Barcode of Life project initiated at the University of Guelph, which allows small samples of tissue to be used to identify the species that the sample came from. For example, a CBC investigative report (CBC News, 2010) found that fish being sold in grocery stores was often being mislabelled; to reach this conclusion, the Marketplace reporters sent samples of the fish meat to be analysed. The samples' barcode regions (the section of the COX1 gene) were sequenced, then this information was compared to the database of known barcode sequences to identify the fish species that the meat actually came from.
This is the last part of the lab assignment. If you have any difficulty with the Clustal instructions (PDF), try viewing the video instructions demonstrating how to use the web resources. Click on the screencapture below to view the video.
CBC News. 2010 Apr 2. Mislabelling means rare fish sold: Marketplace. CBCnews [Internet]. [cited 2011 Dec 20]; Available from: http://www.cbc.ca/news/story/2010/04/01/consumer-fish-marketplace.html